Pancreatic cancer has a higher mortality rate than almost any other malignancy, in large part because most patients receive a diagnosis only after the disease has spread. The five-year relative survival rate stands at 13%, and current systemic therapies offer limited long-term benefit once metastasis is established. A central driver of metastatic spread is WASF3, a member of the Wiskott-Aldrich syndrome protein family that assembles into a heteropentameric WASF Regulatory Complex, WRC, together with ABI1/2, NCKAP1, CYFIP1/2, and BRK1. When activated, the WRC remodels the actin cytoskeleton, generating lamellipodia and invadopodia that propel cancer cells into surrounding tissue. WASF3 upregulation in pancreatic cancer correlates with poor prognosis, heightened invasion, and increased metastasis, yet it was not previously tested whether peptide-based WRC disruption could suppress these phenotypes in a pancreatic setting.
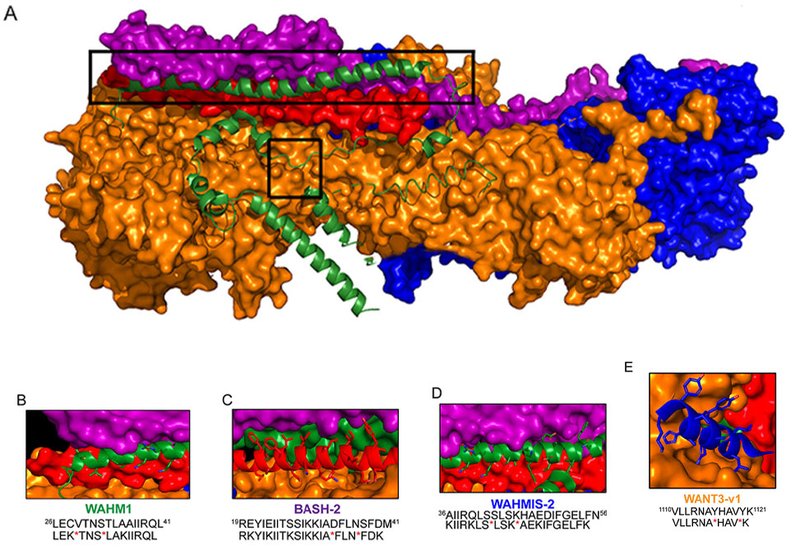
Researchers in the Kennedy Group at The University of North Carolina at Chapel Hill, published in Biochemistry, screened a library of hydrocarbon-stapled peptides previously characterized in breast and prostate cancer models to determine whether they could also suppress WASF3-driven behavior in pancreatic cancer cells. Each constrained peptide mimics a discrete protein-protein interface within the WRC: WAHM1 targets the WASF3-CYFIP1-ABI2 interface, BASH-2 targets the BRK1-WASF3-ABI2 interface, WAHMIS-2 targets a second WASF3 interface, and WANT3-v1 targets the NCKAP1-CYFIP1-BRK1 interface. Primary screening used wound healing and transwell invasion assays in PANC-1 and BxPC-3 pancreatic ductal adenocarcinoma cell lines.
In the initial screen across PANC-1 cells, WAHM1 produced the largest suppression of wound closure, reducing it by 40–50% relative to the DMSO control, while WAHMIS-2 and BASH-2 each achieved roughly 30% inhibition. WANT3-v1 showed no measurable effect in either pancreatic cell line, in contrast to its activity in breast cancer models, suggesting that the relative abundance of individual WRC subunits varies across cancer backgrounds and governs which interfaces are functionally relevant. In BxPC-3 cells, which express lower levels of WASF3 and the WRC than PANC-1, WAHM1 suppressed wound closure by approximately 70% at the same concentration. Dose-response studies in PANC-1 confirmed a concentration-dependent relationship across 1–15 μM, reaching up to 70% suppression of wound healing and 70% inhibition of transwell invasion at 15 μM. BxPC-3 cells responded at lower concentrations, with up to 90% inhibition of wound closure at 10 μM. Critically, MTS viability assays over 72 hours and a cell proliferation assay measuring total DNA content at 24 hours showed no significant reduction in either metric, confirming that the anti-migratory effects of WAHM1 were not attributable to cytotoxicity or growth arrest.
Mechanistic studies in PANC-1 cells established that WAHM1 reaches its intended intracellular targets. Fluorescence microscopy of cells treated with fluorescein-labeled WAHM1 for four hours revealed diffuse cytoplasmic distribution, and flow cytometry demonstrated a time-dependent increase in cellular fluorescence intensity. Streptavidin pulldown with biotin-labeled WAHM1 confirmed direct binding to ABI2 and CYFIP1 within cell lysates; no interaction with WASF3 itself was detected, consistent with the peptide being derived from the WASF3 sequence. Western blotting after a six-hour treatment showed that WAHM1 reduced WASF3 protein levels by approximately 40%, with additional reductions in ABI2 and CYFIP1, a pattern consistent with proteasome-mediated degradation of destabilized WRC subunits. Phalloidin staining revealed a concomitant shift in actin architecture: WAHM1-treated cells displayed dense cytoplasmic stress fibers, whereas DMSO-treated controls showed actin enrichment at the leading edge and no fiber formation, a phenotype indicative of suppressed lamellipodia-driven motility.
These findings establish proof of concept that WRC disruption via stapled peptides can suppress WASF3-driven invasion and migration in pancreatic cancer, extending a strategy previously validated in breast and prostate cancer models into a malignancy with particularly poor therapeutic options. The differential activity of WANT3-v1 across cancer types opens a broader question about context-dependent WRC architecture and which protein-protein interfaces warrant targeting in a given tumor background. Combining WAHM1 with standard chemotherapy or emerging immunotherapies could provide an adjunct strategy to reduce metastatic burden while first-line regimens are active, a potentially meaningful extension of the treatment window for patients with advanced pancreatic disease.